
This is generally made directly on blood smears, or following culture in synthetic medium. When the parasite is not abundant, one can concentrate the parasites first by centrifugation; they are found in the "buffy coat" or white cell layer. The real problem is that the parasite is not abundant in the blood during the indeterminate and chronic stages. This method probably can only detect less than 1% of chronic infections.
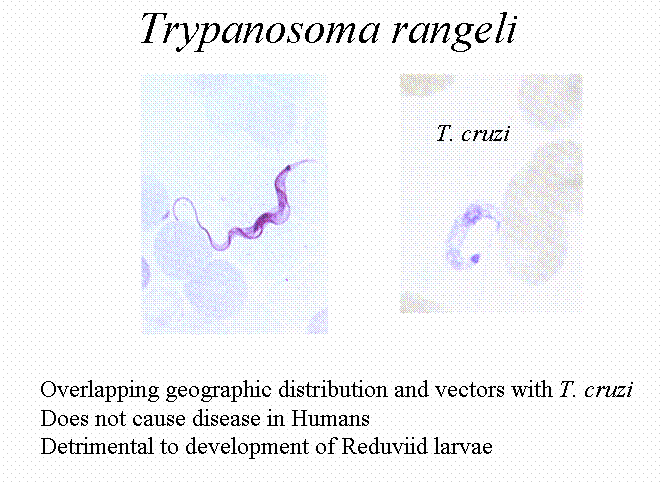
Another problem is that Trypanosoma rangeli looks very much like T. cruzi and can lead to misdiagnosis. T. rangeli is a trypanosome that is infective for humans and other animals but is nonpathogeneic to humans (but detrimental to the insect!). The chief insect host is R. prolixus. The trypomastigotes are discharged in the saliva rather than the feces. It looks morphologically like T. cruzi and has an overlapping geographical distribution.
Xenodiagnosis is a method of diagnosis first described by Alexander Joseph Emile Brumpt (1877-1915). Laboratory-reared non-infected triatomines are allowed to feed on patients suspected to have Chagas disease. It is not painful, but the jar is usually covered in foil so as not to arouse anxiety in the patient. The bugs are then examined 3-4 weeks later for the presence of T. cruzi in the hind gut/excreta; T. rangeli will be found in the salivary glands. Although this method is quite efficient in diagnosing the acute disease, it may be only 50% efficient in the chronic stage. Whereas 1 µl of blood can be viewed (with patience) on a microscope slide, 10 triatomines can sample 1 ml of blood. The efficiency of this technique is complicated by variable growth of different T. cruzi isolates in different genera and species of reduviids. This method has a sensitivity of approximately 50%.

The uninfected bugs are in a vial covered
with gauze and strapped onto the arm of the patient.
These techniques are more sensitive, but in some cases are subject to false positives due to cross-reactivity of antigens common to other trypanosomatids. Serology can detect nearly 100% of chronically infected patients, but false positives with Leishmania and T. rangeli occur.
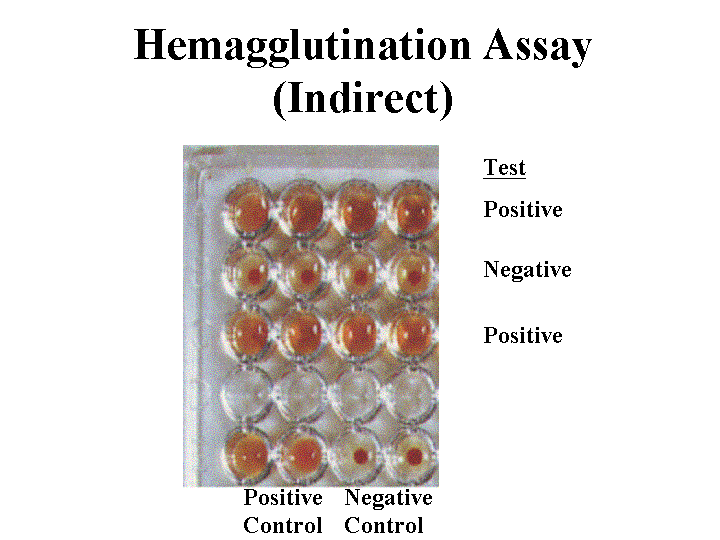
Hemagglutination Assay
Gives results in a few hours. Antigen is bound to glass slide, which is incubated with serum. Anti-T. cruzi antibody is then bound by a fluorescent-labeled anti-constant region antibody. Visualized by microscopy. Detects IgG and IgM antibodies. Can be performed with spots of blood on filter paper (antibody eluted later) - i.e. good for field studies.
Gives results in a few hours. Again, specificity is determined by the quality of antigen. Mucin extracts give good selectivity in a recently-reported assay. Very sensitive assay. Improvements include changing from an immunoperoxidase reaction to a chemiluminescent reaction.
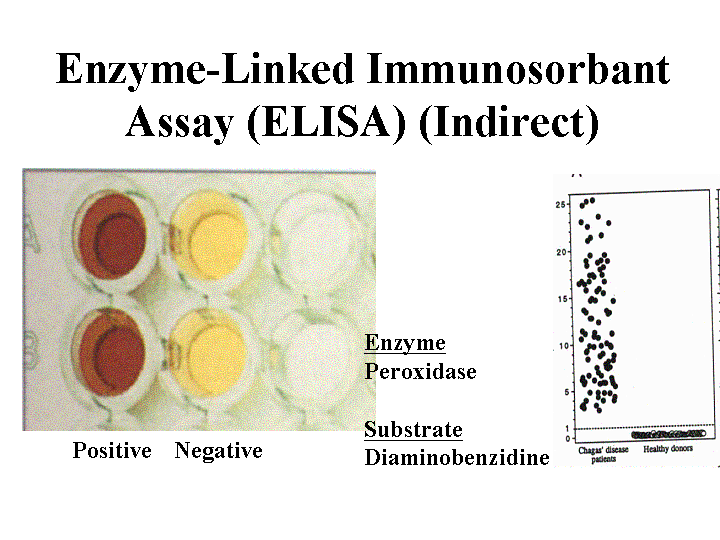
ELISA assays using crude parasite antigens give a high percentage of inconclusive results and false positives. A recent study using two recombinant antigens, CRA (cytoplasmic repetitive antigen) and FRA (flagellar repetitive antigen), gave a reduction in false negatives, in inconclusive results and cross reactions. The assay uses 50 ul serum samples and takes two hr to perform. The assay shows 100% specificity and a sensitivity of 98.2% (110 of 112 chagasic patients).
There are two major criteria for successful PCR detection of a parasite (or any microorganism):
Specificity -- must detect only the T. cruzi parasite
and
Sensitivity -- how few parasites can the assay detect?
In 1989, these criteria were fulfilled for T. cruzi by several gene targets that represent multicopy kinetoplast and nuclear DNA. Both of these assays can detect at least 10 organisms in 100 µl of blood.
Kinetoplast DNA minicircles are catenated together into a single giant network situated in the single mitochondrion of the cell. There are about 10,000 minicircles per network. Each minicircle has four copies of a conserved region that represents about 40,000 copies of the annealing target.
There are multiple minicircle sequence classes defined by the sequence of the variable regions. Morel et al. (1980) showed that high resolution gel analysis of restriction enzyme digested kinetoplast DNA yielded a complex pattern of bands and that these patterns varied between different isolates of T. cruzi. They coined the term, schizodeme, to indicate a strain of T. cruzi that has similar kDNA restriction patterns.
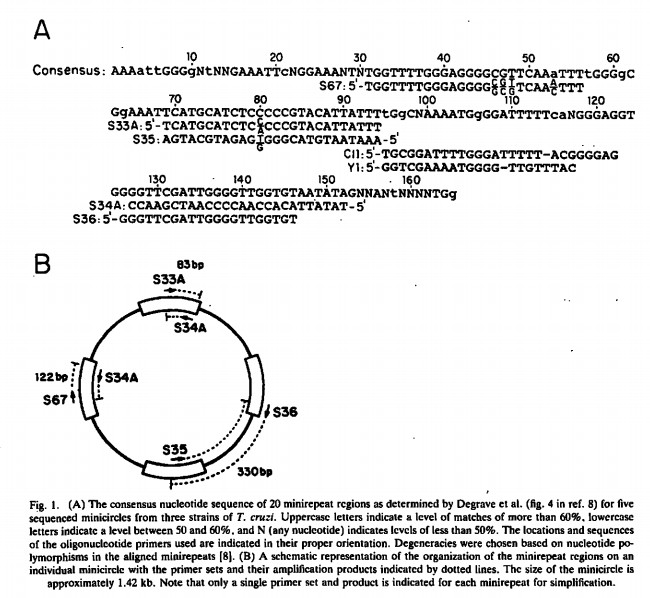

Sturm et
al. (1989) showed that fragments of minicircles could be amplified
using primers within the conserved regions. The Figure above shows the
primers used to obtain an 83 bp, 120 bp or 300 bp product. The 300 bp
product contains the variable regions of all the minicircles and
therefore contains strain information also.
The amplification worked equally well with kDNA from several T. cruzi strains and did not occur with kDNA from other kinetoplastids. The lower limit was approximately 10 trypanosomes. A billion fold excess of human DNA had no effect on the PCR.
The kDNA PCR method was used on a panel of 114 blood samples from chronic Chagasic patients by Avila et al. (1993) and shown to have a sensitivity of 100%. In addition all of the serology-positive and xenodiagnosis negative samples were PCR-positive. Improvements to the method involved the collection of blood in a guanidine-EDTA reagent that lysed the cells and preserved the DNA without refrigeration, cleavage of all minicircles in the network by a chemical nuclease and the use of non-radioactive visualization technology. An example of positive PCR results for the amplified 330 bp fragment from patients' bloods. From Avila et al. (1993).
Recent improvements to this method including the use of boiling the blood lysate instead of chemical nuclease digestion to cleave minicircle DNA have led to the ability to detect 0.1 fg 330 bp DNA (a single parasite cell has 15 fg DNA). Quantitative PCR using the amplification of kDNA minicircle fragments was also used to detect 3-260 parasites per ml of infected rat blood, and in one chronic human patient, 4 parasites per ml of blood were detected.
Click here to see an article in pdf format about the development of this assay.
This method allowed the detection of one parasite in 10 ml of blood. A comparison of the sensitivity of the PCR method and microscopic methods for detecting T. cruzi in blood of infected mice was performed by Kirchoff et al. (1996). They showed that in the acute phase, parasites were detected 3.9 days earlier by PCR than by microscopy. In the chronic phase, the mice were consistently positive by PCR but parasites were only detected intermittently by microscopy.
The major problems with the use of PCR detection of T. cruzi include the cost, the need for specialized equipment, and the frequency of contamination with previously amplified products, which produces false positives.
Isoenzyme Analysis
In addition to detecting the organism, detection assays may be used to distinguish among subgroups of the parasite. One method that has been used extensively is to identify the isoenzyme patterns of the organism.
The groups identified by this methods are called "zymodemes".

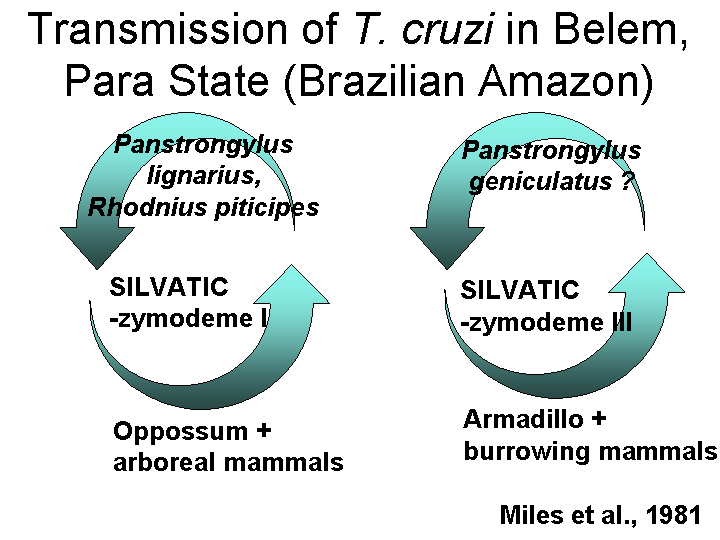
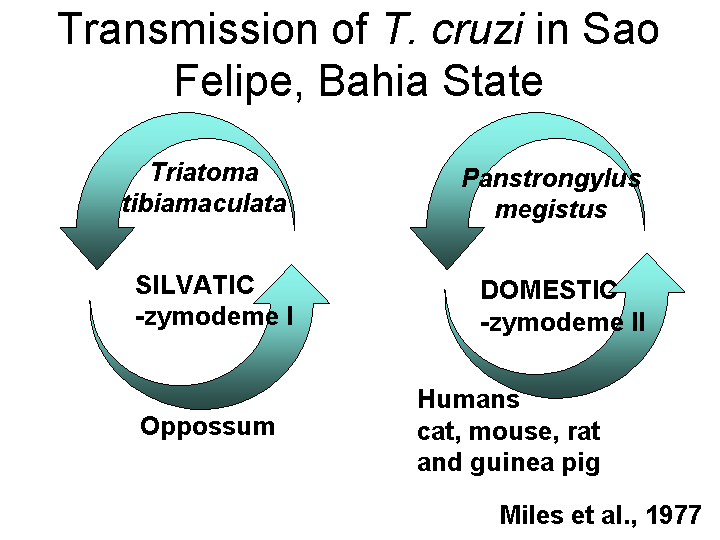


In 1985, Michel Tibayrenc extended these studies and concluded that there were at least 53 different zymodemes (subsequently termed "clonets"). Statistical analysis of these data showed that they did not fit the Hardy-Weinberg Equilibrium model, suggesting that these diploid cells did not have a sexual cycle. A CLONAL Theory of protozoa was proposed - that they replicate only by binary fission.
By the late 1990s, the wheel had turned again and it now seems that there are 3-6 relevant subdivisions within the taxon depending on the assay used: three from analysis of the Spliced Leader RNA gene; at least four from analysis of the ribosomal RNA gene; and six from RAPD (Random Amplified Polymorphic DNA) studies.
The spliced leader or min-exon genes represent a good target for a PCR-based detection method. This RNA is involved in the trans-splicing of mRNA, a process which is found only in kinetoplastid and euglenoid protozoa. There are over 200 tandem repeats each containing a highly conserved 39-nt exon sequence, a moderately variable 55-101 nt transcribed intron sequence, and a highly variable 250-1350 nt nontranscribed intergenic region.
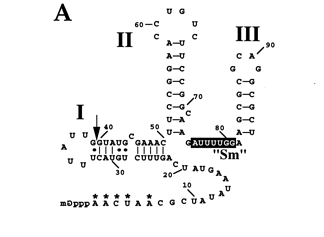
Secondary structure of the L. tarentolae SL RNA based on the L. collosoma structure (form II) that predominates in vivo (29, 44); numbering is relative to the start of transcription. The stem-loop structures are labeled I, II, and III, and the exon-intron junction is indicated by the arrow after nt 39. The Sm-binding-site sequence AUUUUGG is indicated. The 7meG cap is shown at the 5' end, along with the methylated nucleotides (*) that comprise the cap 4 structure (21, 55). From Sturm et al. (1999)
The length and sequence of the repeat was shown to vary between different groups of trypanosomatids (Hassan et al., 1993; Fernandes et al., 1994; Murthy et al., 1992). Marfurt et al. (2003) developed a method to distinguish Old World and New World leishmania by PCR of the spliced leader genes:


The mini-exon repeat of Leishmania.
Each repeat contains a highly conserved exon (39 bp), a moderately
variable transcribed intron region (55 to 101 bp), and a highly
variable non-transcribed spacer sequence (51 to 341 bp). Location of
primer sites (Fme and Rme) are indicated with arrows.
The use of PCR amplification of the mini-exon repeats from a variety of kinetoplastids with a single conserved primer set combined with the detection of specific PCR products by capture hybridization primers specific for the intronic and intergenic regions of the repeats of each species was shown by Ramos et al. (1996).
PCR amplification of the spliced leader genes from different strains of T. cruzi suggested the existence of a distinct sublineage. Use of the Spliced Leader RNA gene as the target can distinguish three groups within T. cruzi (Fernandes et al. (1998): The PCR reaction contains four different oligonucleotides and is known as Multiplex PCR. Addition of a fifth oligonucleotide can also distinguish T. rangeli.
This assay is good for diagnosis (specificity) but not necessarily detection, as the sensitivity is not so good: 100 copies of the gene per cell.
Biological Significance of this Genetic Variation
Clearly, the questions that need to answered are
(1) what is the significance for this genetic variation to an
understanding of the biology of T. cruzi?
(2) Can we predict the eventual outcome of chronic Chagas disease
from knowing the genotype of the parasite? (3) Can we understand why
some reduviid species transmit some isolates of T. cruzi
better than others?
The original Miles results on domestic and sylvatic strains of T. cruzi were extended by Fernandes et al. and Zingales et al. in 1997 using the mini-exon gene marker.
The Zymodeme II appears to also be correlated with parasitemia and pathology, but this is not surprising since it is the domestic strain.
Evolution of T. cruzi
The ancestral trypanosomes probably evolved when there was a single large land mass.
When Africa separated from America, this produced a biological separation of the trypanosomes.
Questions:
1. Distinguish between direct and indirect methods of detection.
2. Xenodiagnosis is considered the "gold standard" for diagnosis of Chagas Disease. Why?
3. Why is PCR possibly a better method than Xenodiagnosis? What are the possible problems with this method?
4. Why might the classification of T. cruzi strains prove to be important in terms of treatment of the disease?